Intro
NOTE: THIS DOCUMENTATION IS FOR STARGAZER_V2.0 If you are using an older version, please download the latest version.
Stargazer is a set of utilities that first began as a single-purpose tool for genotyping pharmacogenetics (PGx) genes from next-generation sequencing (NGS) data. It has now expanded to include the capabilities to genotype PGx genes from both NGS data and single nucleotide polymorphism (SNP) array data, as well as provide comprehensive end-to-end genotyping solutions. Stargazer can also create a genotype data file (GDF) from a BAM/CRAM file.
You can get help on running Stargazer in the console by entering:
python3 stargazer --help
The files / arguments determine the method for which Stargazer will be run. For performing structural variation detection, you must provide both
'-c/--control-gene' and '-g/--gdf-file'. Without the control gene and gdf file Stargazer will run in the VCF-only mode. To create a GDF using Stargazer run as follows:
python3 stargazer –create-gdf-file <name_for_GDF> –control-gene <Control gene> –bam-file <path_to_BAM>
For more information, refer to the extensive documentation.
Usage
Description
Stargazer v2.0 has been simplified to a standalone program one to run as one of three of the commonly used functions of the original Stargazer: genotyping with and without the addition of read depth information, and the generation of the required file (GDF) for use when attempting to call star alleles with copy number or structural variant contributions. In this section, we will first explain how Stargazer's genotyping algorithm works using the CYP2D6 gene as an example. After that, we will provide some example usages and the full list of command-line arguments.
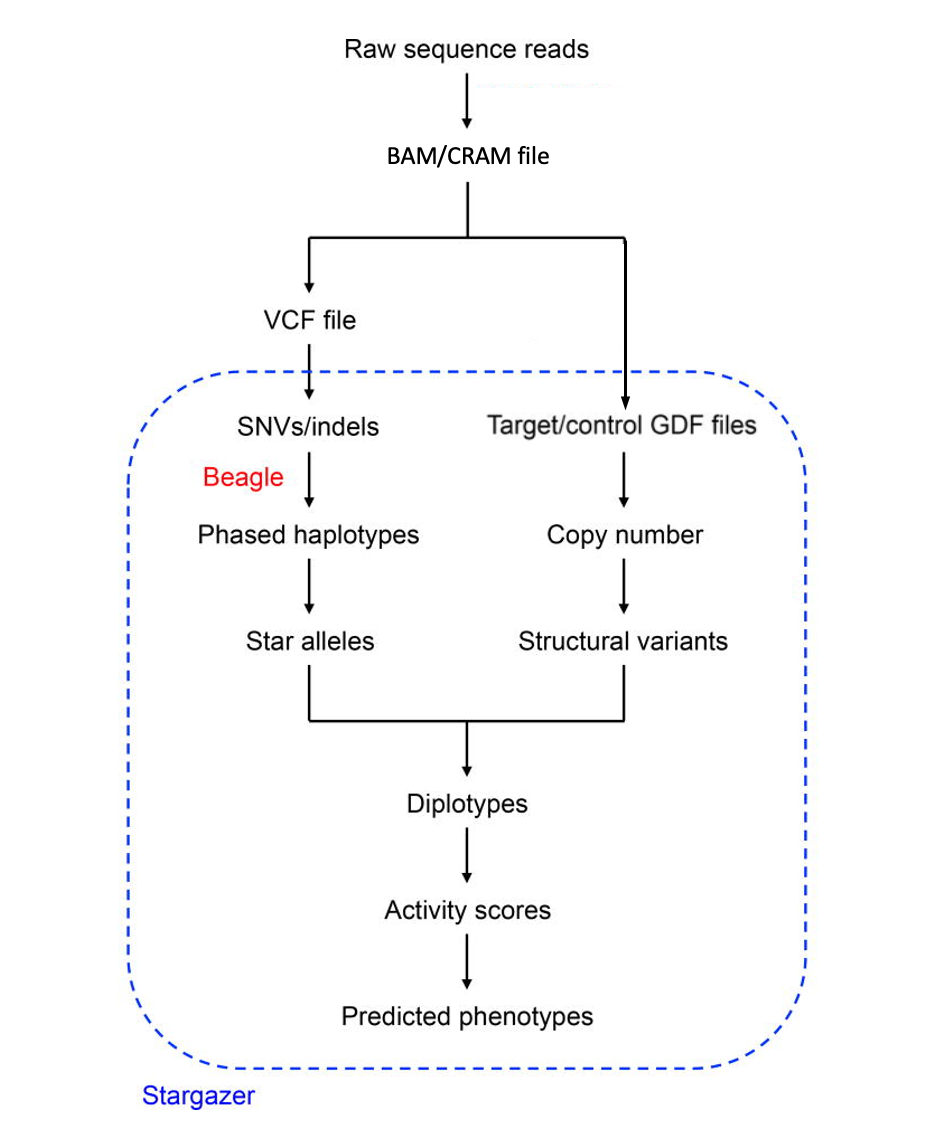
Figure 1. A schematic diagram of the Stargazer genotyping pipeline for any star allele, like many on CYP2D6, that requires read depth information for the determination of copy number (CNV) of other structural variant (SV) contributions to the genotype. For star alleles that are defined solely with single-nucleotide (SNV) or small insertion/deletion (indel) variants, the left path flow using a VCF file as input is all that is required. This figure was modified from an original taken from Lee et al., 2018.
Part I. Input and output data of Stargazer
The Stargazer CYP2D6 genotyping pipeline is outlined in Figure 1. The pipeline uses BAM- or CRAM-formatted files comprised of sequence reads aligned with a mapping program (such as BWA-MEM or DRAGEN) to either the GRCh37 or GRCh38 human reference genome assembly. From these BAM or CRAM files, the user must initially generate a VCF file (again, with variant-calling software such at GATK-HaplotypeCaller or DRAGEN) from which Stargazer extracts all SNVs/indels located within 3kb from either end of CYP2D6. More specifically, Stargazer stores the genomic position of each variant, the reference allele, the alternate allele(s), the genotype status (homozygous or heterozygous), and the allelic depth for each sample. Stargazer uses the variant information from the VCF file to call star alleles based on SNVs/indels.
BAM/CRAM files are also used to calculate read depth for CYP2D6 and CYP2D7 with a new internal feature of Stargazer. For convenience, we will continue to refer to this output with the GDF (GATK-DepthOfCoverage format) file. Using the -G/–-create-gdf-file as shown below, Stargazer v2.0 uses a few internal Python routines to generate the necessary files containing information of read depth across both the gene of interest (target gene) and a known user-chosen copy-number 2 reference gene (control gene) for use as a read depth normalization factor; with both sets of information being contained in one GDF file. Since the high homology between CYP2D6 and CYP2D7 can cause reads to align to erroneous or multiple locations, only uniquely mapping reads with a mapping quality >= 20 are counted. Stargazer computes paralog-specific copy number using read depth information from the target and control genes in the GDF files in order to detect SVs.
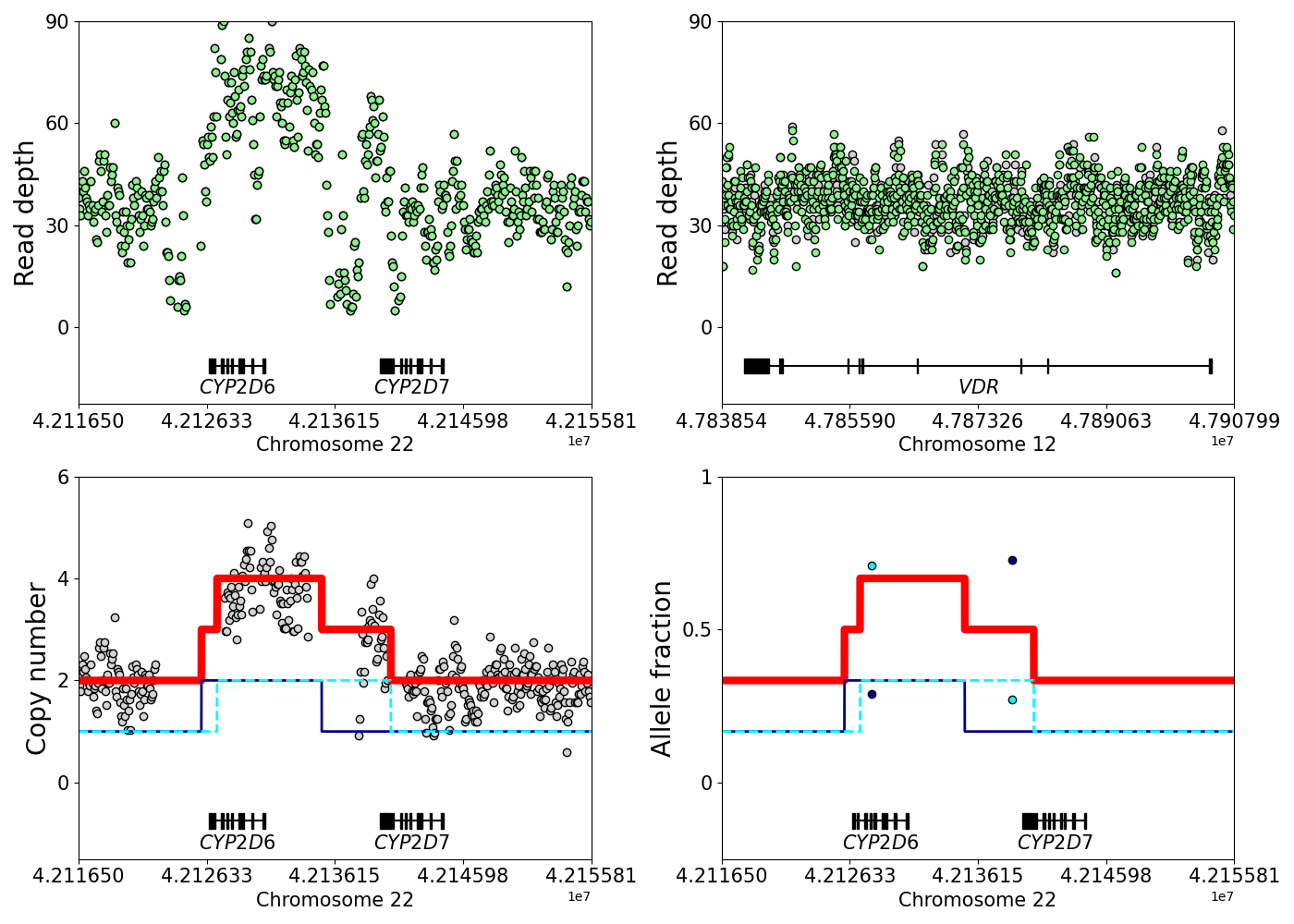
The output data of Stargazer include each sample's CYP2D6 diplotype, predicted phenotype, and plots to visually inspect copy number for CYP2D6 and CYP2D7 (Figure 2). Stargazer also, in a finalized.vcf file, returns all detected SNVs/indels including those that are novel as well as those that are known but are not currently used to define any star allele, so investigators will know which variants to potentially follow up. In addition, those variants can be annotated with SeattleSeq Annotation and used to define new star alleles. Note that when calling diplotypes, Stargazer only considers those variants that are currently used by the Pharmacogene Variation Consortium (PharmVar).
Figure 2. An example of a hybrid star allele detected by Stargazer: CYP2D6 *2x2/*68+*4 diplotype. (*68 is a CYP2D6-CYP2D7 hybrid with a switch region in intron 1; it is found in tandem with *4.) Green dots are points of raw read depth, shown here for both the target and control genes. Grey dots are copy number calculated from read depth. The light and dark blue dots show the allele fraction values of relevant SNV or indel variants.
Part II. Prediction of star alleles
From a VCF file, Stargazer uses Beagle to haplotype phase heterozygous variants for CYP2D6 with over 2,500 reference samples from the 1000 Genomes Project. Stargazer then matches phased haplotypes to star alleles with a translation table built from publicly available data (e.g., the Pharmacogene Variation Consortium). The table contains information on more than 90 star alleles and 185 SNVs/indels including variant positions and nucleotide changes in relation to the reference CYP2D6*1 allele and the GRCh37 and GRCh38 human reference genome assemblies.
Part III. Detection of SVs
From a GDF file, Stargazer converts read depth for CYP2D6 and CYP2D7 (target gene) to copy number by performing intra- and inter-sample normalizations. The former accounts for individual variation in sequencing efficiency using read depth from a control gene; also contained in the GDF file, while the latter considers heterogeneity in coverage across all samples. Stargazer automates the detection of SVs using four of the most basic Python modules: numpy, pandas, scipy and matplotlib that approximate one or more points at which the statistical properties of a sequence of observations change. Here, the sequence is DNA, the observation is per-base copy number, and the statistical property is the mean copy number. If there is a significant shift in the mean copy number (e.g., from 2 to 1), the algorithm returns the change point location and the two mean values (e.g., 2 and 1).
Part IV. Identification of diplotypes
For samples without SVs, Stargazer determines CYP2D6 diplotypes by combining the star allele used to assign each phased haplotype. For samples with a whole gene deletion, the affected haplotype is assigned the CYP2D6*5 deletion allele, which is then combined with the star allele assigned to the other haplotype to form a diplotype. For samples with a whole gene duplication, the affected haplotype will be assigned "x2" (e.g., CYP2D6*1x2, *2x2, etc.) because it has two gene copies of CYP2D6. For samples with more complex SVs such as CYP2D6/CYP2D7 hybrids, individual algorithms have been developed to determine diplotypes.
Part V. Assignment of predicted phenotypes
There are four CYP2D6 metabolizer classes: poor, intermediate, normal, and ultrarapid. To predict these phenotypes, Stargazer first translates CYP2D6 diplotypes into a standard unit of enzyme activity known as an activity score. The fully functional reference CYP2D6*1 allele is assigned a value of 1, decreased function alleles such as CYP2D6*10 and *17 receive a value of 0.5, and non-functional alleles including CYP2D6*4 and *5 have a value of 0. The sum of values assigned to both alleles constitutes the activity score of a diplotype. Consequently, subjects with CYP2D6*1/*1, *1/*4 and *4/*5 diplotypes will have an activity score of 2, 1 and 0, respectively. These activity scores are used to predict the four metabolizer classes as following: poor, 0; intermediate, 0.5; normal, 1 to 2; ultrarapid, greater than 2.
Part VI. Reporting
Stargazer v2.0 creates a report that shows the phased haplotypes as well as other possible candidates to consider such as unphased, multiple star alleles in one haplotype, and warns against BEAGLE imputed missing genotypes.
Main usages
Genotype with WGS data (or TS data)
stargazer -a <genome_build> -o <output_prefix> -d wgs (or -d ts) -t <target_gene> -i <vcf> -c <control_gene> -g <gdf>
Genotype with WGS-generated (or TS-generated) VCF only
stargazer -a <genome_build> -o <output_prefix> -d wgs (or -d ts) -t <target_gene> -i <vcf>
Genotype with SNP array-generated VCF only
stargazer -a <genome_build> -o <output_prefix> -d chip t <target_gene> -i <vcf>
Genotype with SNP array-generated VCF only, using the Beagle program's imputation algorithm
stargazer -a <genome_build> -o <output_prefix> -d chip t <target_gene> -i <vcf>
-impute
Command-line arguments
|
Options |
Argument |
Description |
|
-v/--version |
. |
Print the Stargazer version number and exit. |
|
-i/--vcf-file |
PATH |
Path to the input VCF file (.vcf or .vcf.gz). An empty VCF file containing only the header and metadata is still accepted by Stargazer as it means no variants were detected from the sample(s). [required for standard running] |
|
-o/--output-dir |
PATH |
Path to the output directory. If the directory with the same name already exists Stargazer will overwrite it. [required for all processes] |
|
-t/--target-gene |
TEXT |
Name of the target gene. [required for all processes] |
|
-a/--genome-build |
TEXT |
Build of the reference genome assembly used to create the input data. [default: 'hg19'] |
|
-d/--data-type |
TEXT |
Type of the input data: wgs whole genome sequencing; ts targeted sequencing including whole exome sequencing; or chip single nucleotide polymorphism microarray. [default: 'wgs'] |
|
-c/--control-gene |
TEXT |
Name of a preselected control gene. Used for intrasample normalization during copy number analysis. Alternatively you can provide a custom genomic region with the 'chr:start-end' format (e.g. chr12:48232319-48301814). [required with --create-gdf-file] |
|
-g/--gdf-file |
PATH |
Path to a GDF file containing read depth for both the target and control genes. |
|
-r/--hap-panel |
PATH |
Path to a reference haplotype panel file (.vcf or .vcf.gz). By default Stargazer uses the haplotype panel from the 1000 Genomes Project (phase 3). |
|
-s/--sample-list |
TEXT |
List of space-delimited sample IDs to be used as control for intersample normalization during copy number analysis. This argument has an effect only when the input data type is targeted sequencing. |
|
-p/--include-profiles |
. |
Use this tag to include profiles for read depth copy number and allele fraction. This tag has no effect if Stargazer is running in the VCF-only mode. |
|
-b/--impute-ungenotyped-markers |
. |
Use this tag to impute ungenotyped markers during statistical phasing. Potentially useful for low-density DNA microarray data. |
|
-st/--star_table |
PATH |
Use this tag to specify the path for star_table to be used [default=f {program_dir}/star_table.tsv] |
|
-G/--create-gdf-file |
TEXT |
This tag causes Stargazer to run as a GDF-file generator. Argument requires a file name (which will be placed in the output directory). [must include -B option] |
|
-B/--bam-file |
TEXT |
List of paths to BAM or CRAM files. [required for --create-gdf-file runs] |
|
|
|
*IS BAM LIST TO BE SPACE-DELIMITED? |
Interpretation of results
Here's the explanation of the column headers in output_prefix.stargazer-genotype.txt files:
|
Header |
Description |
|
name |
Sample ID used in the VCF file and the GDF file |
|
status |
Genotype status with following choices: g (genotyped), ng (not genotyped) |
|
hap1_main |
Main star allele for the 1st haplotype |
|
hap2_main |
Main star allele for the 2nd haplotype |
|
hap1_cand |
Candidate star alleles for the 1st haplotype |
|
hap2_cand |
Candidate star alleles for the 2nd haplotype |
|
hap1_score |
Activity score for the 1st haplotype |
|
hap2_score |
Activity score for the 2nd haplotype |
|
dip_score |
Combined activity score for both haplotypes |
|
phenotype |
Predicted phenotype based on the diplotype activity score |
|
dip_sv |
Combination of two unphased SV calls |
|
hap1_sv |
SV call for the 1st haplotype |
|
hap2_sv |
SV call for the 2nd haplotype |
|
ssr |
The sum of squared residuals (SSR) computed during the SV detection step |
|
dip_cand |
All possible candidate star alleles, obtained by combining the SNPs of both haplotypes to create a pseudo-haplotype |
|
hap1_main_core |
Core SNPs of the 1st haplotype's main star allele |
|
hap2_main_core |
Core SNPs of the 2nd haplotype's main star allele |
|
hap1_main_tag |
Tag SNPs of the 1st haplotype's main star allele |
|
hap2_main_tag |
Tag SNPs of the 2nd haplotype's main star allele |
|
hap1_af_mean_gene |
Mean allele fraction for the 1st haplotype, calculated using all detected SNPs from the target gene |
|
hap2_af_mean_gene |
Mean allele fraction for the 2nd haplotype, calculated using all detected SNPs from the target gene/td> |
|
hap1_af_mean_main |
Mean allele fraction for the 1st haplotype, calculated using the SNPs of the main star allele |
|
hap2_af_mean_main |
Mean allele fraction for the 2nd haplotype, calculated using the SNPs of the main star allele
|
Here's the explanation of the column headers in report.txt files:
|
Name |
Description |
|
Sample |
name of the sample |
|
Gene |
name of the gene |
|
Diplotype |
diplotype (hap1_main/hap2_main) |
|
BEAGLE imputed |
if hap1_main or hap2_main is imputed that star-allele will be listed |
|
Phenotype |
the phenotype calculated as per the additive activity score of the diplotype |
|
Score |
the additive activity score calculated for the diplotype |
|
May also be |
the other possible candidates of haplotypes as per the alleles present in the sample |
|
Also possible haplotype |
the other possible candidates of haplotypes as per the alleles present in the sample if not phased |